


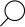
Wykrywanie fenyloketonurii – jak wyglądają badania przesiewowe u noworodków?
Fenyloketonuria (ang. Phenylketonuria - PKU) jest chorobą wrodzoną, dziedziczną w sposób autosomalny recesywny. Wynika z zaburzenia pośredniej przemiany aminokwasu aromatycznego- fenyloalaniny do tyrozyny. Do powstania choroby przyczynia się mutacja genu PAH zlokalizowanego w 12 chromosomie.
Fenyloketonuria, jako choroba dziedziczna
Fenyloketonuria jest metaboliczną chorobą genetyczną. Dziecko może urodzić się chore, gdy zarówno ojciec, jak i matka cierpią na tę chorobę lub są jej nosicielami. Prawdopodobieństwo jej wystąpienia w przypadku choroby dwojga rodziców wynosi 25%. Natomiast, jeśli fenyloketonuria dotyka tylko jednego z rodziców, wówczas dziecko w żadnym wypadku nie odziedziczy schorzenia.
Ta jednostka chorobowa polega na zaburzeniach przemiany fenyloalaniny i nadmiernym jej gromadzeniu się. Fenyloalanina to jedno z białek występujących w organizmie każdego człowieka. Fenyloketonuria jest chorobą bardzo niebezpieczną, nieleczona może prowadzić do poważnego uszkodzenia ośrodkowego układu nerwowego. Choroby nie da się całkowicie wyleczyć, ale można zapobiec jej rozwojowi - jeżeli zostanie zdiagnozowana na odpowiednim etapie.
Fenyloketonuria a badania przesiewowe u noworodków
Ministerstwo Zdrowia wprowadziło program badań przesiewowych noworodków realizowany przez Instytut Matki i Dziecka. Podstawą prawną jest art. 48 Ustawy z dnia 27 sierpnia 2004 r. o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych. Ostatnie dostępne dane z programu pochodzą z lat 2015-2018. Dzięki takim badaniom szybciej można zdiagnozować chorobę u noworodków, których w Polsce jest ok. 380 tys., by wcześnie rozpocząć ich leczenie i profilaktykę.
W Polsce od 1965 roku wszystkie noworodki poddawane są badaniom przesiewowym w kierunku najczęściej występujących chorób metabolicznych, w tym właśnie fenyloketonurii. Badanie to polega na pobraniu krwi dziecka na specjalną bibułkę, a rozpoznanie choroby potwierdza się na podstawie oznaczenia ilości fenyloalaniny we krwi noworodka. Jej szybkie wykrycie i rozpoczęcie leczenia pozwalają uniknąć nieodwracalnego uszkodzenia ośrodkowego układu nerwowego.
POLECANE PRODUKTY:



Koszt badań przesiewowych u noworodków
Rodzice nie muszą się martwic o cenę badań przesiewowych noworodków, gdyż są to badania obowiązkowe i bezpłatne - finansowane z budżetu państwa. W Polsce badania te obejmują 29 jednostek chorobowych. To plasuje nas na dość wysokim poziomie w UE – tuż obok krajów skandynawskich.
Z kolei w Europie Zachodniej (Niemcy, Francja, Hiszpania, Wielka Brytania) są to już badania dotyczące grupy 20 i mniej jednostek chorobowych. Z kolei w USA rekomenduje się 34, a można wykonać badania przesiewowe noworodków do nawet 55 chorób. Ministerstwo Zdrowia w Polsce na badania przesiewowe noworodków w latach 2015-2018 wydało 11 564 593,50 zł. Koszt wykrycia 1 chorego dziecka to 42 206,55 zł.
Sposoby diagnostyki fenyloketonurii
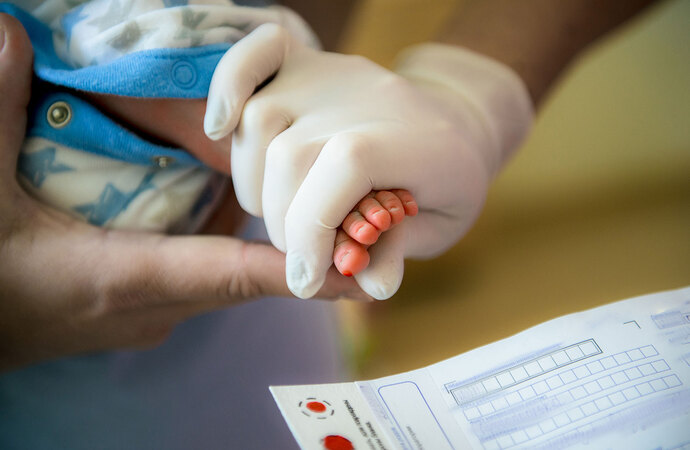
Podstawowe badanie to testy przesiewowe przeprowadzone w 3 dobie życia . Gdy wynik stężenie fenyloalaniny nie przekracza 3 mg/dl, choroba zostaje wykluczona. Jeżeli jest podwyższone stężenia fenyloalaniny(≥ 8 mg/dl) to dziecko zostaje kierowany do ośrodka specjalistycznego celem przeprowadzenia dalszej diagnostyki. Wynik pośredni (3–8 mg/dl) wymaga powtórzenia badania.
Potwierdzenie diagnozy to wykonanie badań genetycznych mających na celu stwierdzenie mutacji genu hydroksylazy fenyloalaninowej w postaci delecji (ubytku) fragmentu genu.
Rodzaje fenyloketonurii
Przyjmuje się podział choroby w zależności od wyjściowego stężenia fenyloalaniny w okresie noworodkowym (według aktualnych wartości European PKU Guidelines:
- stężenie Phe > 20 mg/dL (>1200µmol/l) – klasyczna fenyloketonuria
- stężenie Phe 15-20 mg/dL (900-1200µmol/l) – umiarkowana fenyloketonuria
- stężenie Phe 10-15mg/dL (600-900 µmol/l) – łagodna fenyloketonuria
- stężenie Phe > 20 mg/dL (>1200µmol/l) – łagodna hiperfenyloalaninemia
Stan dziecka z fenyloketonurią zaraz po narodzinach właściwie nie różni się niczym od stanu charakteryzującego zdrowe dziecko. U wcześniaków lekarze obowiązkowo sprawdzają występowanie tzw. wyników fałszywie dodatnich. Powodem ich występowania nie jest jedynie nieskuteczność testu, ale również brak dojrzałości układu enzymatycznego.
Po urodzeniu poziom fenyloalaniny we krwi jest w normie, ponieważ zostaje on wyrównany w okresie płodowym przez enzym matki, a dziecko uznawane jest za zdrowe. Jednakże należy bacznie obserwować i sukcesywnie badać noworodka, ponieważ w ciągu kilku dni stężenie fenyloalaniny, w przypadku chorego dziecka, wzrasta do poziomu, który znacząco przekracza normę.
Do wystąpienia pierwszych objawów choroby dochodzi około trzeciego miesiąca życia. Z biegiem czasu zaczynają się one stopniowo nasilać. W przypadku około połowy osób chorych obserwuje się zmiany skórne o typie łojotokowym czy też takie przypominające wyprysk alergiczny. Nadmiar fenyloalaniny uszkadza przede wszystkim układ nerwowy dziecka. Brak leczenia powoduje pogłębianie się zaburzeń neurologicznych. Osoby chore charakteryzuje dość typowa cecha tzw. „rozcieńczenie barwnika” - czyli występowanie jasnej karnacji w połączeniu z włosami w kolorze blondu czy niebieskich tęczówek.

Charakterystyczne objawy fenyloketonurii u noworodków
- charakterystyczny tzw. mysi zapach wydzielany przez malucha,
- drgawki i drżenie kończyn,
- czasem zdarzają się wymioty,
- sporadycznie występować może wysypka na skórze.
Wraz z postępem choroby, pojawiają się często inne zaburzenia, takie jak:
- zaburzenia rozwoju psychoruchowego u dziecka,
- wady chodu i postawy,
- zachowania dziecka podobne do autystycznych, np. strach przed innymi osobami,
- problemy ze snem,
- agresja wobec siebie i innych ludzi,
- nieuzasadnione oraz niekontrolowane napady złości,
- psychoza.
Bardzo ważna jest wczesna diagnoza, gdyż postęp choroby jest szybki i powoduje nieodwracalne zmiany w organizmie. Bardzo istotne jest wprowadzenie od razu po zdiagnozowaniu odpowiedniej diety. To jedyny środek łagodzenia objawów choroby, gdyż nie ma na nią żadnego lekarstwa. Jeśli fenyloketonuria zostanie odkryta w pierwszych tygodniach życia i natychmiast zostanie wdrożony odpowiedni sposób odżywiania, dziecko będzie prawidłowo się rozwijać.
Po wykryciu choroby u dziecka, należy w ciągu maksymalnie 2 tygodni od urodzenia wprowadzić zbilansowaną dietę eliminacyjną, czyli tzw. niskofenyloalaninową. Przy takiej diecie stosuje się specjalne mieszanki, które zawierają niską zawartość białka oraz zawarty w nim aminokwas zwany fenyloalaniną.
Ile fenyloalaniny można spożywać?
To bardzo indywidualna kwestia. Zależna od specyfiki pacjenta, rodzaju fenyloketonurii oraz występowania innych chorób. Z kolei u dzieci nieznaczące ilości fenyloalaniny są niezbędne do ich prawidłowego rozwoju. Jak dotąd formalnie i oficjalnie nie określono konkretnej dawki spożycia fenyloalaniny, która byłaby bezpieczna dla chorych na fenyloketonurię – podobnie jak w przypadku glutenu.
By dokonać prawidłowej oceny, ile fenyloalaniny pacjent może spożyć, niezbędne jest regularne i częste badanie poziomu tego aminokwasu we krwi. Zazwyczaj dziecko, które nie ukończyło trzeciego roku życia, poddaje się cotygodniowym testom. Następnie co dwa tygodnie, a gdy skończy 7 lat – co miesiąc. Dziecko musi się stosować do zaleceń aż do momentu, gdy mózg zwolni tempo rozwoju, czyli do ukończenia 12 - 14 lat. Potem ograniczenia w żywieniu można nieco zliberalizować. Jednak na ogół zaleca się taką dietę stosować przez całe życie - mniej lub bardziej rygorystycznie.
Podsumowanie
Dzieci, które cierpią na fenyloketonurię, powinny wyeliminować z jadłospisu wszystkie produkty wysokobiałkowe, a więc m.in. mięso, ryby, jaja, mleko, przetwory mleczne, orzechy, kakao, fasolę, soję, wiele produktów mącznych oraz aspartam, który jest źródłem zakazanej fenyloalaniny. Ponadto powinny przyswajać specjalne preparaty PKU, które dostarczają organizmowi niezbędnych witamin i soli mineralnych. Co więcej, na rynku dostępne są także specjalnie opracowane produkty niskobiałkowe PKU. Dzięki nim pacjenci będą w stanie prowadzić w miarę normalną dietę i jeść posiłki, które są alternatywą dla wielu tradycyjnych artykułów. Jednak prowadzenie prawidłowej diety, które umożliwi dziecku prawidła rozwój i funkcjonowanie wymaga stałych i częstych konsultacji z lekarzem oraz dietetykiem.



Skomentuj artykuł